Índice
| 1. | Introducción |
| 2. | Proceso de evaluación de riesgos |
| 3. | Las agencias de evaluación de riesgos en Europa y en EE.UU. |
| 4. | Críticas |
| 5. | Referencias |
- Introducción
Las personas están expuestas a cientos de sustancias químicas cada día. Más de 85.000 productos químicos se utilizan en productos de consumo (Goldman y Koduru 2000) y se sabe que más de 6.000 productos químicos se utilizan en materiales que están en contacto con alimentos. (Oldring y col. 2013). Si la exposición no se controla adecuadamente, las sustancias químicas pueden causar daño a las poblaciones expuestas. Los efectos sobre la salud pueden variar desde la irritación ocular leve y náuseas a enfermedades crónicas graves como el cáncer. Con el fin de controlar la exposición a sustancias químicas, se realizan evaluaciones de riesgos. La evaluación de riesgo químico es la cuantificación del riesgo que tiene la población de sufrir daños en la salud debido a la exposición química. El enfoque básico para cuantificar el riesgo sigue la siguiente fórmula:
| RISK = EXPOSURE x HAZARD |
La evaluación del riesgo debe distinguirse claramente de la gestión del riesgo, que es un proceso normativo llevado a cabo por los reguladores; en la gestión del riesgo se tienen en cuenta las consideraciones socio-económicas o políticas y se sopesan frente a las estimaciones del riesgo. Posteriormente, se inician las medidas de control y prevención que sean necesarias. En general, la evaluación del riesgo se considera un proceso con base científica que consta de cuatro etapas: a) identificación del riesgo, b) evaluación dosis-respuesta (o: cuantificación de riesgos), c) evaluación de la exposición y d) caracterización del riesgo (Figura 1) (NRC 1983).
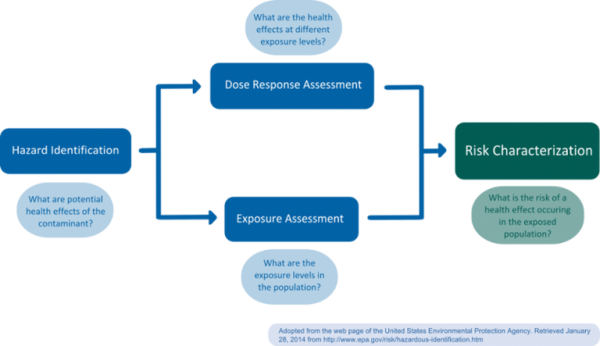
- Proceso de evaluación de riesgos
- a) Identificación de los riesgos
Durante la identificación del peligro se identifica el peligro potencial que plantea la sustancia química. La información que habitualmente se requiere para la identificación de sustancias químicas peligrosas incluye datos sobre toxicocinética (absorción, distribución, metabolismo y eliminación (ADME)) y la interacción de una sustancia química con una diana biológica (toxicodinámica, por ejemplo, las relaciones cuantitativas estructura-actividad (QSAR) (EPA 2014). Otras fuentes alternativas de información son los accidentes y los niveles de residuos medidos en la biota (van Leeuwen y Vermeire 2007).
- b) Evaluación dosis- respuesta
El objetivo de la evaluación de dosis-respuesta es describir la relación entre los diferentes grados de exposición (dosis) a una sustancia identificada como un peligro potencial, y la incidencia y la gravedad de un efecto sobre el organismo, centrándose normalmente en la diana más sensible (van Leeuwen y Vermeire 2007). Las evaluaciones dosis-respuesta se basan generalmente en estudios toxicológicos in vivo, aunque también se pueden emplear estudios epidemiológicos in vitro, in silico. Los estudios in vivo para la regulación de la evaluación del riesgo se llevan a cabo normalmente de acuerdo con las directrices establecidas por la Organización para la Cooperación y el Desarrollo Económico (OCDE), que siguen las Buenas Prácticas de Laboratorio (BPL). Como resultado de la evaluación dosis-respuesta se estableció una ingesta diaria tolerable (IDT o Dosis de Referencia (DR) en los EE.UU.) para la población humana de interés. Para las sustancias no cancerígenas, el TDI o dosis de referencia corresponde a un nivel de exposición en el que no se prevé ningún efecto perjudicial para la salud (dosis segura). Para extrapolar el estudio en animales a la población humana, el nivel de efecto adverso no observado (NOEL o NOAEL, nivel en el que no se observó ningún efecto en un estudio en animales) por lo general, se divide por un factor de seguridad de 100 (van Leeuwen y Vermeire 2007). En los casos en los que no haya un NOAEL disponible, se puede utilizar límites estadísticos inferiores tales como el Índice de Referencia Nivel de Dosis (BMDL) para estimar los niveles de dosis seguros (Crump 2003). Para los compuestos sin umbral, como los carcinógenos genotóxicos, el TDI o dosis de referencia se refiere a un nivel de riesgo aceptable que se fija por lo general a 1/1.000.000. Por lo general, el nivel de confianza inferior al 95% se utiliza como punto de partida para la extrapolación; la extrapolación puede basarse en el modo-de-acción, o puede ser lineal (EPA 2005).
- c) Evaluación de la exposición
Durante la evaluación de la exposición, se estiman los niveles de exposición encontrados o, el en caso de productos químicos que aún no están comercializados, los niveles que se prevé que se van a dar en la población. Para este propósito se puede utilizar la biomonitorización o los modelos de exposición. Los métodos de biomonitorización incluyen la medición de los niveles en orina o suero en un estudio representativo de la población. Los modelos de exposición requieren la estimación de las cantidades de sustancia química liberadas, de las vías y de los mecanismos de transformación o degradación (van Leeuwen y Vermeire 2007). En el ámbito de los materiales en contacto con alimentos (FCM), los modelos de exposición combinan a menudo encuestas alimentarias con distribuciones del mercado de envasado y datos sobre migración. Un modelo reciente basado en este enfoque es la herramienta FACET (Flavours, Additives and food Contact material Exposure), sobre sabores, aditivos y materiales en contacto con los alimentos (FACET, Oldring y col. 2013). Se ha sugerido también la monitorización de la Planta de Tratamiento de Aguas Residuales (WWTP, Waste water treatment plant) como un indicador de la exposición química de las personas (Venkatesan y Halden 2014) y fue utilizado anteriormente para calcular la exposición a productos químicos perfluorados (PFC) (D’eon y col. 2009).
- d) Evaluación del riesgo
En esta última etapa los evaluadores del riesgo integran la información recogida durante los pasos anteriores para cuantificar la probabilidad real, «el riesgo», de un efecto perjudicial sobre la población expuesta (cociente de riesgo, van Leeuwen y Vermeire 2007). Se incluye información adicional acerca de las suposiciones e incertidumbres de los pasos anteriores. La caracterización del riesgo puede diferenciar por sexo, edad, estilo de vida y/o grupos étnicos, para permitir una comprensión más diversificada de los riesgos que tiene un producto químico de causar daños dentro de una población. La caracterización del riesgo debe adherirse a los principios de transparencia, claridad, coherencia y razonabilidad (TCCR, EPA 2014).
- Las agencias de evaluación del riesgo en Europa y en EEUU
En Europa, la evaluación del riesgo está formalmente separada de la gestión del riesgo. Mientras que la Autoridad Europea de Seguridad Alimentaria (European Food Safety Authority, EFSA) se encarga de realizar una evaluación con base científica del riesgo de los FCM, la Comisión Europea, en particular, el Directorate General for Consumer Health (DG SANCO), el Parlamento Europeo y los estados miembros de la UE, se encargan de integrar la evaluación científica del riesgo con las consideraciones de carácter político, económico, social y medioambiental (EFSA 2014). En los EEUU, la “Food and Drug Authority” (FDA) se encarga tanto de la evaluación como de la gestión del riesgo. Tanto en los EE.UU. como en Europa, el riesgo derivado de sustancias que no están reguladas específicamente o que están exentas de regulación, también debe ser evaluado de forma independiente por los fabricantes.
- Críticas
- – No siempre existe una clara distinción entre la gestión del riesgo «subjetiva» y la evaluación del riesgo «objetiva»; la evaluación del riesgo también requiere decisiones normativas (van Leeuwen y Vermeire 2007).
- – No queda claramente definido el concepto de «riesgo aceptable» para la exposición de la población y los efectos no cancerígenos sobre la salud (Martin y col. 2013).
- – Los factores estándar de seguridad se establecen de forma un tanto arbitraria y no siempre representan los peores escenarios (Falk-Filipsson y col. 2007, Hasegawa y col. 2010, Martin y col. 2013).
- – La evaluación dosis-respuesta no siempre cubre suficientemente los riesgos de las dosis bajas (exposiciones reales) (Crump 2003).
- – Los modelos utilizados en la evaluación de la dosis-respuesta no están científicamente fundados (dicotomía de mecanismos lineales y no lineales, efectos de bajas dosis) (Crump 2003)
- – Los estudios in vivo a la escala habitual sólo permiten detectar niveles de riesgo (el riesgo de desarrollar un cierto criterio de valoración de la enfermedad) por encima del 1% de frecuencia en las poblaciones de roedores; los niveles de riesgo ambiental de interés normativo son frecuentemente muy inferiores (por ejemplo 1 en 100.000 (Crump 2003).
- – La información sobre la liberación, las vías y los mecanismos de transformación o degradación, la exposición humana, así como el metabolismo humano de los productos químicos, es a menudo incompleta y, a veces, está asociada a grandes incertidumbres (van Leeuwen y Vermeire 2007)
- – El riesgo no siempre aumenta con el aumento de la exposición (curvas dosis-respuesta no monótonas para los productos químicos que imitan hormonas, Zoeller y col. 2012)
- – No toda incertidumbre es cuantificable (en el sentido de que se pueda calcular un «riesgo»); la incertidumbre restante puede ser cualitativa y no cuantificable (Bradley y Drechsler, 2013)
- Referencias
Bradley, R. and Drechsler, M. (2013). “Types of Uncertainty.” Erkenntnis, 1-24.
D’Eon, J.C., et al. (2009). «Observation of a commercial fluorinated material, the polyfluoroalkyl phosphoric acid diesters, in human sera, wastewater treatment plant sludge, and paper fibers.« Environ Sci Technol 43:4589-4594.
EFSA (2014). “About EFSA.” Retrieved January 29, 2014 from the web page of the European Food Safety Authority.
EPA (2005). “Guidelines for carcinogen risk assessment – EPA/630/P-03/001F.” Retrieved February 11, 2014 from the web page of the US EPA.
EPA (2014). “EPA risk assessment.” Retrieved January 29, 2014 from the web page of the U.S. EPA.
Falk-Filipsson et al. (2007). “Assessment factors—Applications in health risk assessment of chemicals.” Environmental Research 104, 108–127
Goldman, L.R. and Koduru, S. “Chemicals in the environment and developmental toxicity to children: a public health and policy perspective.” Environmental Health Perspectives 108(Suppl 3): 443–448.
Hasegawa, et al. (2010). “Proposal of new uncertainty factor application to derive tolerable daily intake.” Regulatory Toxicology and Pharmacology 58, 237–242.
Martin, O. et al. (2013). “Dispelling urban myths about default uncertainty factors in chemical risk assessment –sufficient protection against mixture effects?”. Environmental Health 12:53.
NRC (National Research Council, 1983). “Risk assessment in the federal government:managing the process.” National Academy Press, Washinton, DC. US.
Oldring, P. et al. (2013). “Development of new modeling tool (FACET) to assess exposure to chemical migrants from food packaging.” Food Additives & Contaminants: Part A.
Venkatesan, A. and Halden, R. (2014). “Wastewater Treatment Plants as Chemical Observatories to Forecast Ecological and Human Health Risks of Manmade Chemicals.” Scientific Reports 4.
Van Leeuwen, C.J. and Vermeire, T.G. (2007). “Part I. Introduction” in Risk assessment of chemicals: an introduction. 2nd edition.” Springer.
Zoeller, T. et al. (2012). “Endocrine-Disrupting Chemicals and Public Health Protection: A Statement of Principles from The Endocrine Society.” Endocrinology 153, 9.
* May be broken down into one safety factor for intra-species variability (factor of 10, aims to account for differences between rodents and humans) and one for inter-species variability (also a factor of 10, animals used for in vivo studies are highly inbred and thus have a very homogenous genetic background which does not reflect natural genetic variability) (Renwick 1991)
